
This is the fifth edition of this visualisation, previous editions were in June 2015, June 2014, October2013 and December 2012.
As before, full run throughput in gigabases (billion bases) is plotted against single-end read length for the different sequencing platforms, both on a log scale. Yes, I know a certain new instrument (different from last time) seems to be missing, hang on, I’m coming back to that…
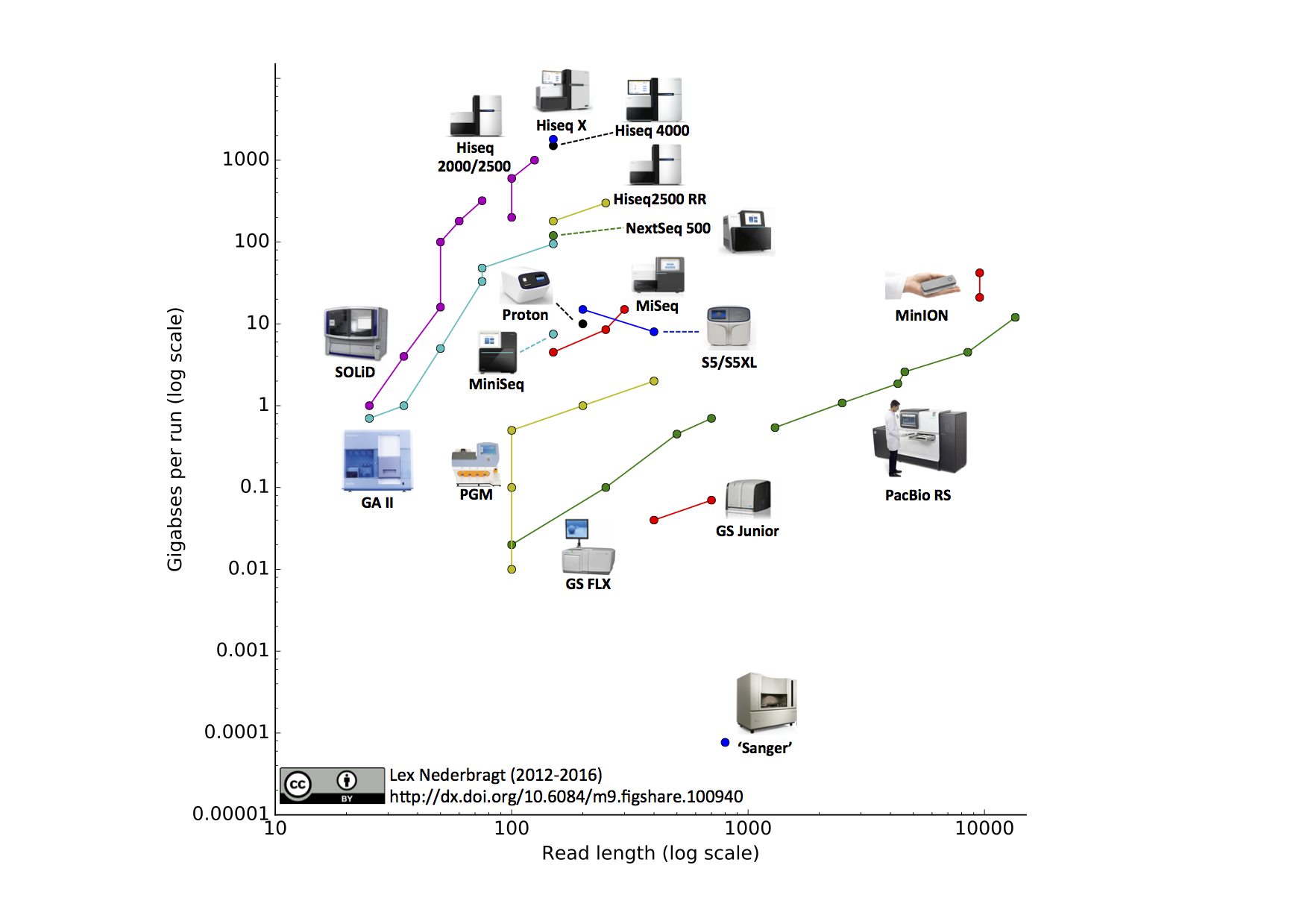
Notable changes from the June 2015 edition
- I added the Illumina MiniSeq
- I added the Oxford Nanopore MinION. The read length for this instrument was based on the specifications for maximal output and number of reads from the company’s website. The two data points represent ‘regular’ and ‘fast’ modes.
- I added the IonTorrent S5 and S5XL. You may notice that the line for this instrument has a downward slope, this is due to the fact that the 400 bp reads are only available on the 520 and 530 chip, but not the higher throughput 540 chip, making the maximum throughput for this read length lower than for the 200 bp reads.



